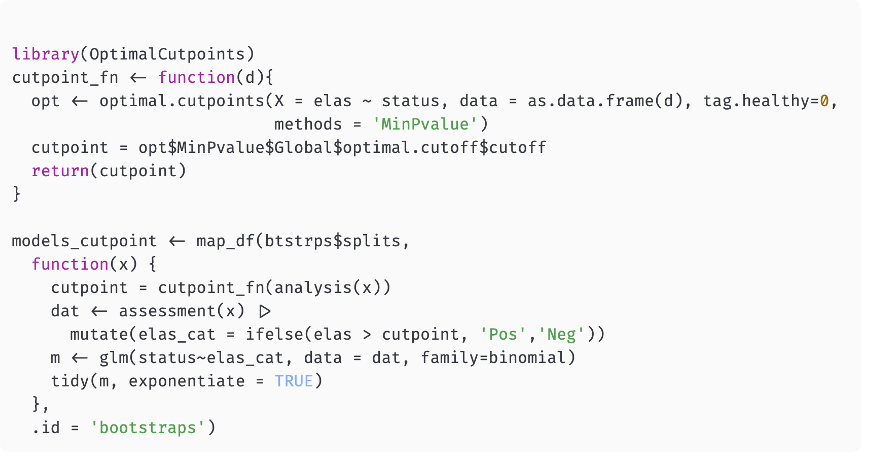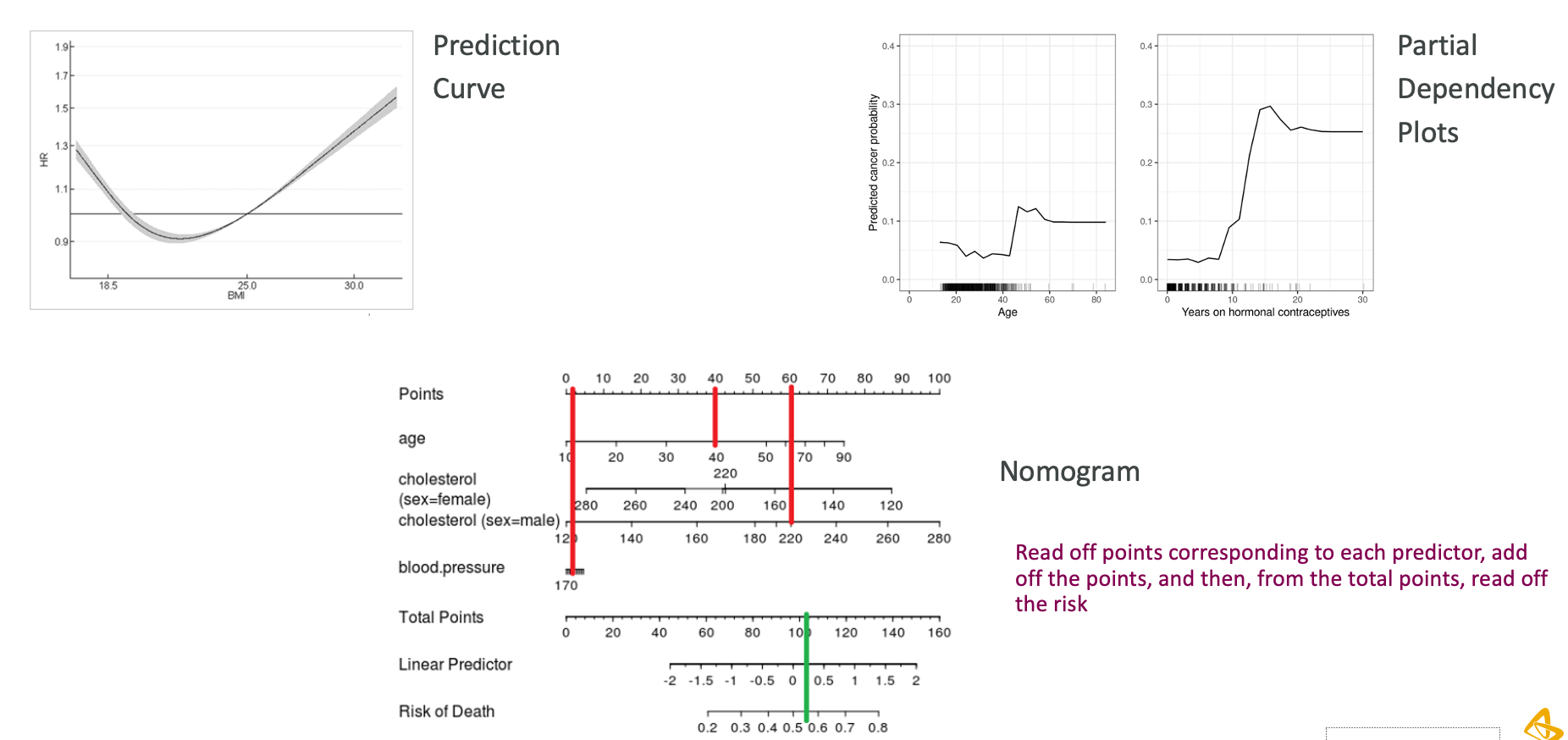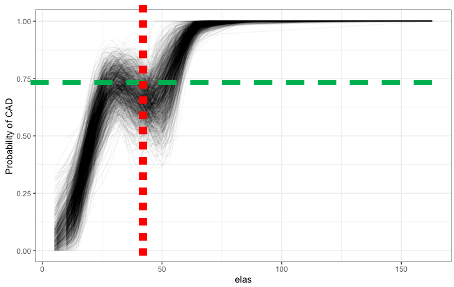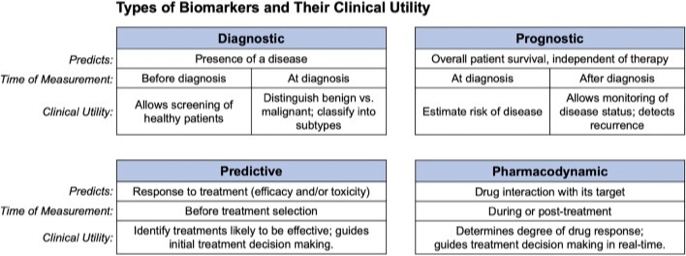









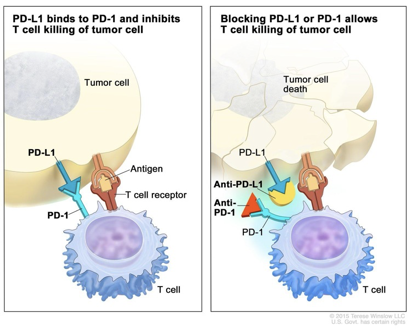

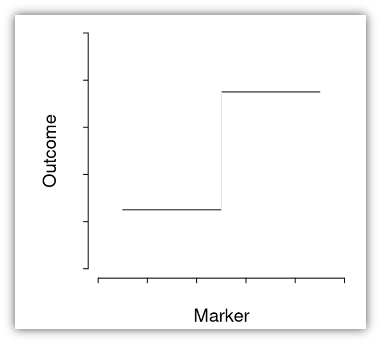

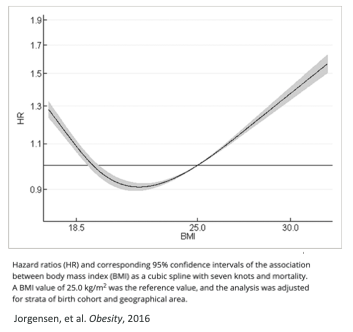
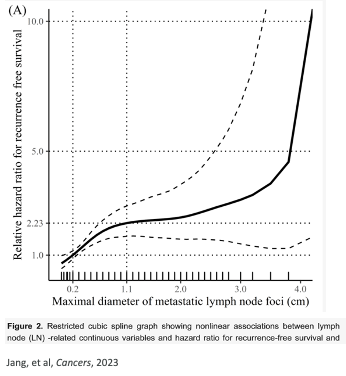



library(tidyverse)
library(tidymodels)
library(rms)
data(elas, package="OptimalCutpoints")
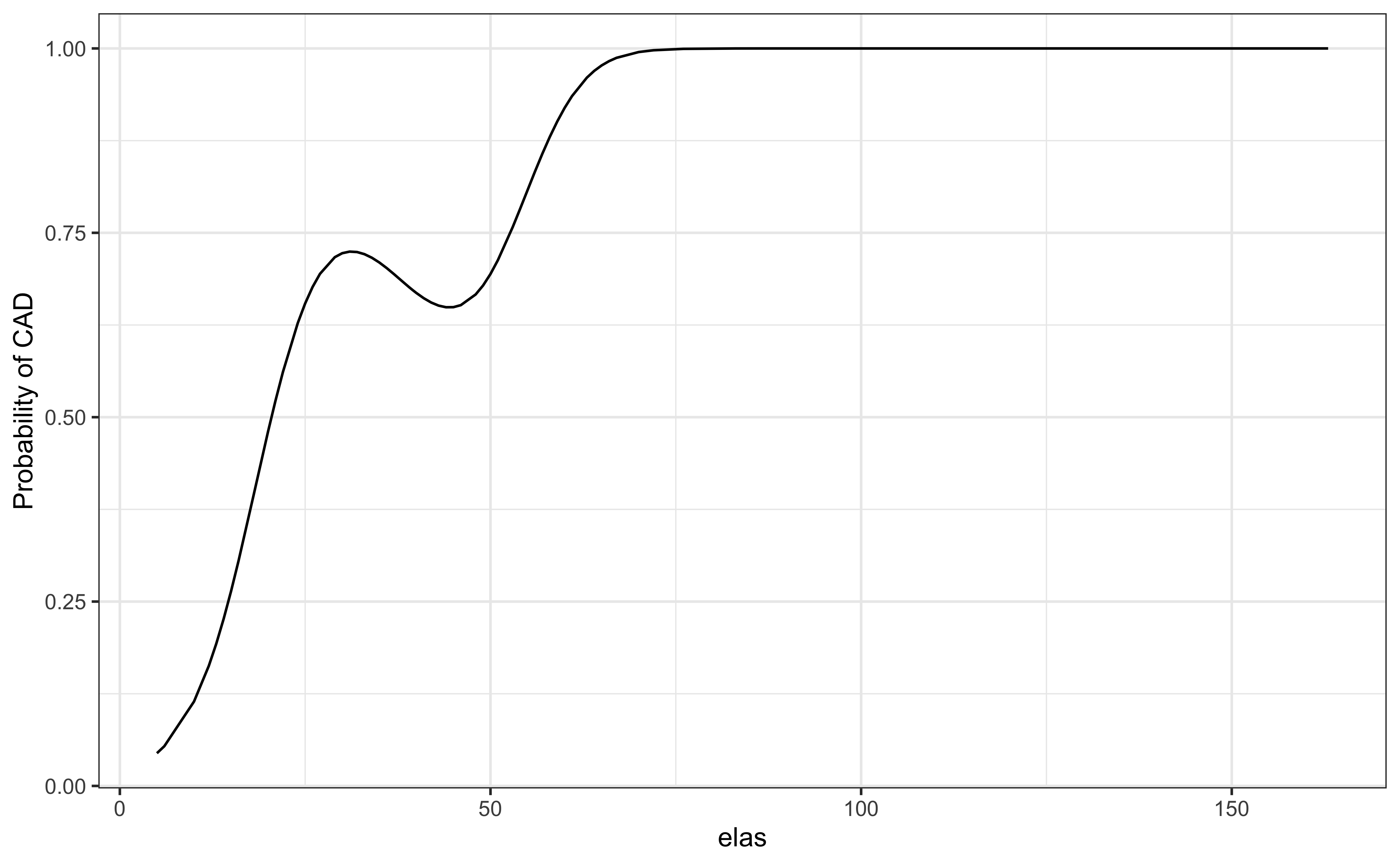
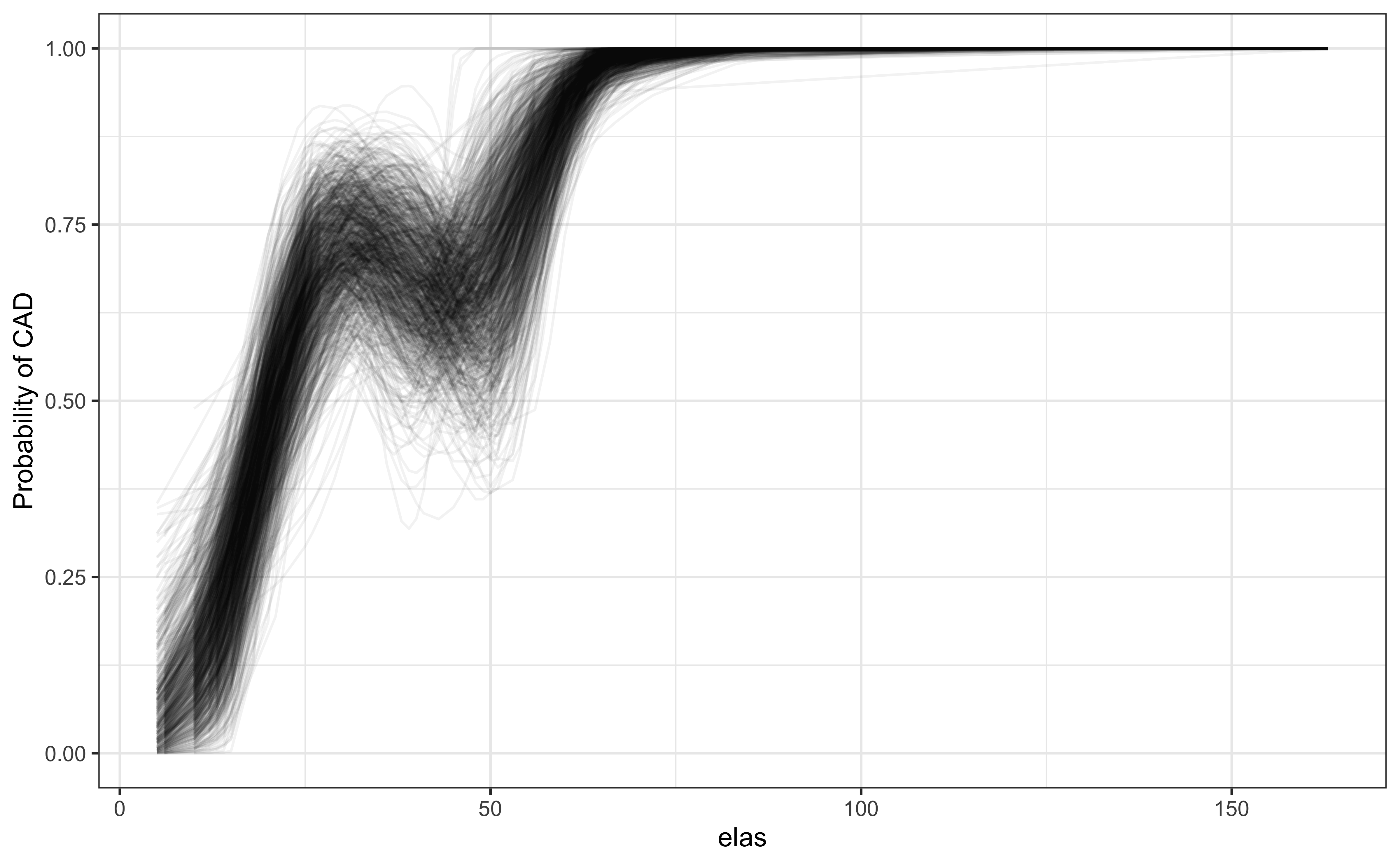
model <- glm(status ~ rcs(elas), data = elas, family = binomial)
a <- augment(model, newdata=elas, type.predict = 'response')
ggplot(a, aes(elas, .fitted))+geom_line()+theme_bw()+
labs(y = "Probability of CAD")